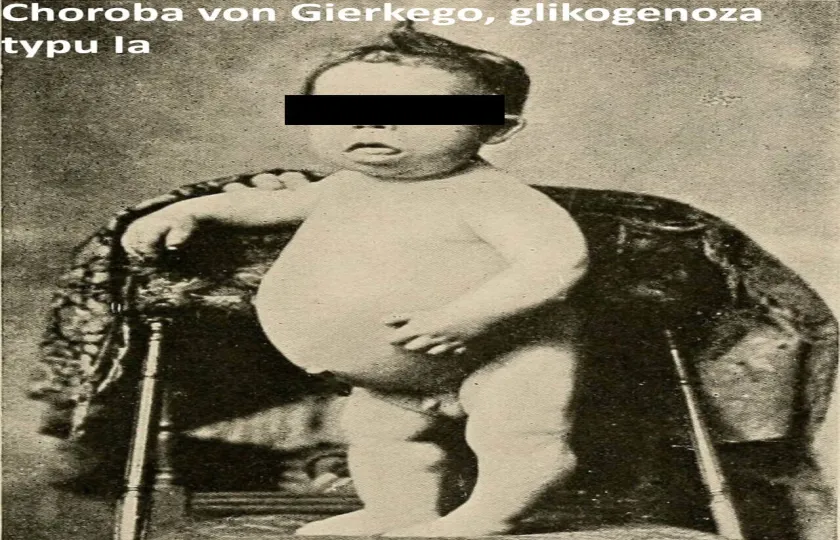
Choroba von Gierkego, glikogenoza typu Ia, glycogen storage disease Ia, von Gierke disease, GSD Ia jest najczęstszą chorobą glikogenną. Ta genetyczna choroba jest spowodowana niedoborem enzymu glukozo-6-fosfatazy, który upośledza zdolność wątroby do tworzenia glukozy poprzez rozkład glikogenu i glukoneogenezę. Ponieważ w wyniku działania tych dwóch mechanizmów wątroba utrzymuje prawidłowy poziom glukozy w celu zaspokojenia wszystkich potrzeb metabolicznych organizmu, przy niedoborze tego enzymu procesy te nie przebiegają prawidłowo, co prowadzi do hipokaliemii.
Naruszenie systemu rozkładu glikogenu powoduje gromadzenie się tej substancji w wątrobie i nerkach, a to odpowiednio prowadzi do zwiększenia objętości tych narządów. Pomimo wzrostu, nerki i wątroba nadal normalnie pełnią swoje funkcje w dzieciństwie, ale w wieku dorosłym stają się podatne na różne zmiany zachodzące w organizmie. Innymi następstwami zaburzeń metabolicznych może być kwasica mleczanowa (gromadzenie się kwasu mlekowego we krwi i tkankach obwodowych) oraz hiperlipidemia. Aby uniknąć tych powikłań, głównym sposobem leczenia jest stałe stosowanie węglowodanów o dużej masie cząsteczkowej, takich jak skrobia kukurydziana lub inne, w celu utrzymania poziomu glukozy poprzez stopniowe wchłanianie glukozy, która powstaje, gdy skrobia jest rozkładana z pożywienia. Aby leczyć inne problemy związane z chorobą Gierkego, potrzebne są inne metody leczenia.
Choroba została nazwana na cześć niemieckiego lekarza Edgar Otto Conrad von Gierke, który jako pierwszy ją opisał.
Enzym glukozo-6-fosfataza znajduje się na wewnętrznej błonie retikulum endoplazmatycznego. Reakcja katalityczna, w której bierze udział ten enzym obejmuje białko wiążące wapń oraz trzy białka transportowe (T1, T2, T3), które ułatwiają przemieszczanie się do centrum katalitycznego odpowiednio glukozo-6-fosforanu (G6P), glukozy i fosforanu w czasie tej reakcji.
Najczęstszymi postaciami HD są typ Ia (80% przypadków) i typ Ib (20% przypadków). Ponadto istnieją inne formy, które są bardzo rzadkie.
Typ Ia wynika z mutacji w genie G6PC kodującym glukozo-6-fosfatazę (G6P). Gen ten znajduje się na 17q21chromosom.
Typ Ib jest spowodowany mutacją w genie T1, Transporter G6P. Cechy metaboliczne typów Ia i Ib są bardzo podobne, ale w przypadku choroby na Ib pacjent może doświadczyć dodatkowych powikłań, a ten typ jest uważany za nieco trudniejszy.
Glikogenoza typu Ia dziedziczona jest w sposób autosomalny recesywny. Heterozygotyczni nosiciele (rodzice) zwykle nie mają żadnych objawów choroby. Podczas gdy prawdopodobieństwo zachorowania dziecka wynosi 25%. Diagnoza prenatalnaprzeprowadzona w 18-22 tygodniu ciąży z biopsją wątroby, ale jak dotąd nie opracowano specyficznego leczenia tego zaburzenia na tym etapie. Ponadto diagnostyka prenatalna jest możliwa na podstawie DNA płodu uzyskanego w wyniku biopsji kosmówki, ale tylko wtedy, gdy wiadomo, że płód jest zagrożony rozwojem tej choroby.